欢迎来到商名企业
商名企业首页
网站导航
铁死亡是一种由大量脂质过氧化物积累所驱动的调节性细胞死亡方式,与肿LIU发病密切相关,其调控机制尚不清楚。因此,深入理解肿LIU细胞抵抗铁死亡的分子机制,将为肿LIU治L 提供新策略。
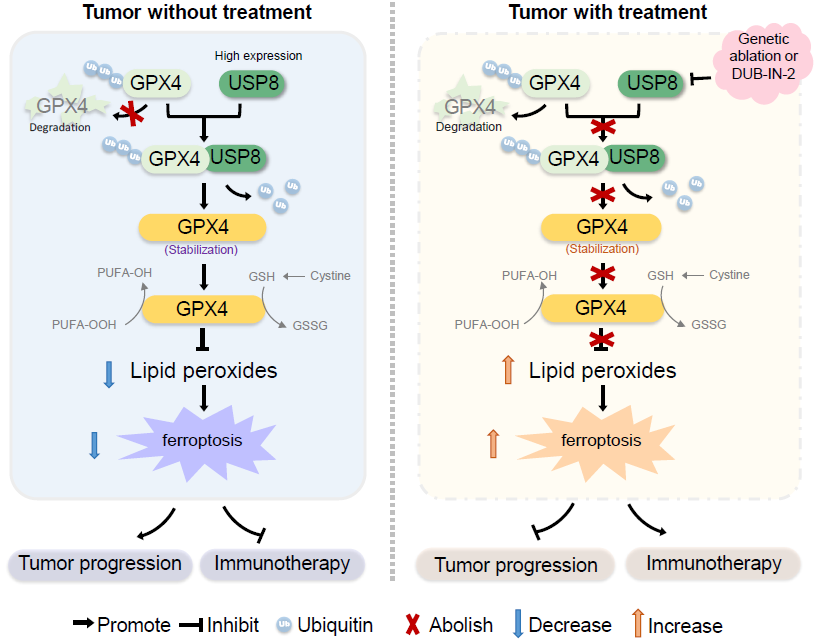
2024年4月,武汉大学医学研究院、教育部免疫与代谢前沿科学中心、中南医院以及泰康生命医学中心团队在PNAS杂志上,发表“USP8-governed GPX4 homeostasis orchestrates ferroptosis and cancer immunotherapy”的研究成果。该研究揭示了USP8通过稳定谷胱甘肽过氧化物酶4(GPX4)来抵抗铁死亡,并强调了靶向USP8作为一种潜在的治L 策略来促进铁死亡,以增强AI症免疫治L 。
图形摘要:
Highlights如下:
1)USP8的特异性敲除导致小鼠过早死亡,并伴有结肠上皮结构紊乱和脂质过氧化迹象。
2)抑制USP8可以增加肿LIU细胞对铁死亡的敏感性。改善PD-1/PD-L1阻断抗体的治L 效果。
3)机制上,USP8与GPX4相互作用,并去除GPX4的多聚泛素化修饰,从而抑制蛋白酶体介导的GPX4蛋白降解,抑制铁死亡,促进肿LIU进展。
表型相关性:
Usp8的特异性敲除导致小鼠寿命缩短,肠上皮细胞死亡增加和脂质过氧化迹象增多。
功能研究:
体外抑制USP8增加肿LIU细胞对铁死亡的敏感性。USP8抑制剂联合铁死亡诱导剂能明显抑制肿LIU生长并改善PD-1/PD-L1阻断抗体的治L 效果。
机制研究:
前面的研究显示USP8能抑制铁死亡。为了进一步探索USP8介导铁死亡调控的分子机制,检测USP8是否可以调节铁死亡通路中关键蛋白的表达。
(1)初步确定下游的蛋白分子
通过WB检测铁死亡相关蛋白,发现敲低明显降低GPX4蛋白的表达(图1a-b),但不影响其RNA水平(图1c)。同样抑制剂亦能降低GPX4的蛋白水平(图1d)。构建GPX4 WT及其酶促失活突变体U46S进行功能实验,证实过表达GPX4 WT蛋白在抑制RSL3诱导的铁死亡方面具有功能(图1e)。放线菌酮(CHX)试验表明,敲低USP8缩短GPX4蛋白的半衰期(图1f)。表明USP8可能作为一种去泛素化酶在翻译后水平调节GPX4的蛋白丰度。蛋白酶体介导的降解系统和自噬-溶酶体系统是调控蛋白质稳态的两大系统。而实验表明蛋白酶体抑制剂MG132挽救了敲减USP8介导的GPX4不稳定,而溶酶体抑制剂巴弗洛霉素A1 (BafA1)和氯喹(CQ)则无此作用(图1g),表明USP8介导的GPX4稳态调节取决于蛋白酶体系统。
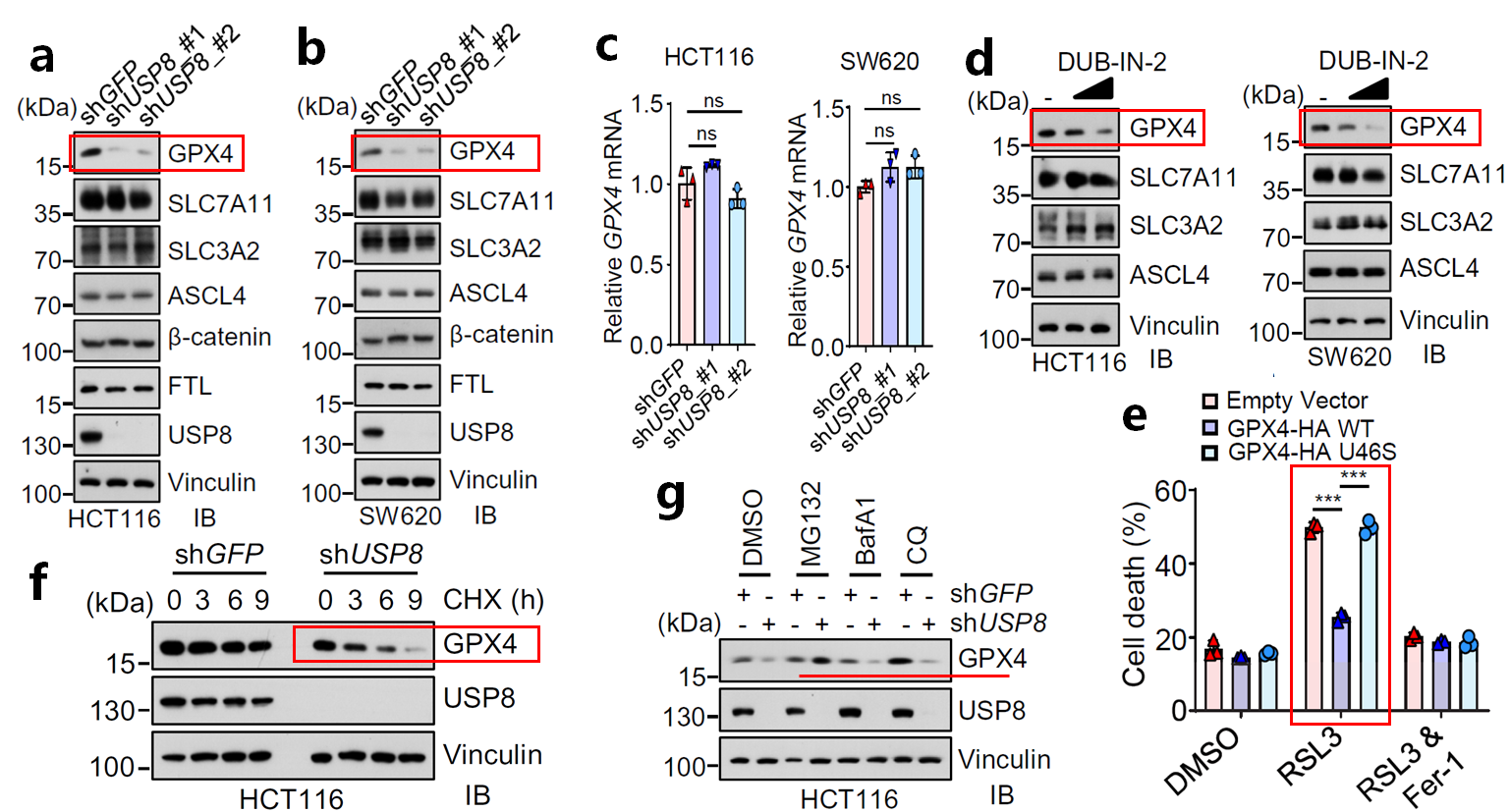
图1 USP8影响GPX4蛋白的稳定性(Ref. Fig 4)
(2)分子互作机制
第一步,确定与下游蛋白分子的互作关系
为了探索USP8与GPX4是否互作,进行内源Co-IP实验和GST pulldown实验,发现USP8与GPX4互作(图2a-c)。
第二步,USP8与GPX4蛋白结合的具体的位置
为了探索USP8与GPX4蛋白结合的位置,针对USP8构建不同的突变体分别进行Co-IP实验,发现USP8通过 aa1-313、aa715-1118区域与GPX4结合(图2d-e)。
第三步,USP8如何影响GPX4蛋白的稳定性
USP8是一个去泛素化酶。Co-IP分析发现,USP8能降低GPX4的泛素化水平,而酶非活性突变体USP8-c786a则不能去除GPX4的泛素链,表明USP8通过其去泛素化酶活性去除GPX4的泛素化(图2f)。

图2 USP8通过其去泛素化酶活性去除GPX4的泛素化(Ref. Fig 4/S4)
第四步,确定USP8去除GPX4的Ub K48链泛素化
众所周知,泛素分子可以通过其七个赖氨酸(K)残基中的一个(K6、K11、K27、K29、K33、K48和K63)连接形成不同的泛素链。基于此,Co-IP分析发现GPX4被K27和K48连接的泛素化修饰(图3a)。另外,Co-IP分析显示USP8过表达降低GPX4上K48连接的泛素化,而不影响K27连接的泛素化(图3b),表明USP8主要去除GPX4上K48连接的泛素化。体外去泛素化试验表明(注:Ub(K48O)(只有完整的Lys48残基的Ub)),USP8过表达以去泛素酶活性依赖性方式去除GPX4上K48连接的泛素化(图3c)。
第五步,确定USP8去除GPX4蛋白泛素化位点
之前的研究已报道K48、K125、K127、K135和K151是GPX4的潜在泛素化位点。构建不同的泛素化位点突变体进行Co-IP分析,发现GPX4 K48R、K125R、K127R和K151R突变体K48连接的泛素化减少(图3d)。另外,USP8过表达降低GPX4 WT、K125R、K127R和K151R突变体的泛素化,但未能消除GPX4 K48R突变体的泛素化(图3e)。表明USP8主要去除GPX4上K48残基的泛素化。
综上所述,这些结果表明USP8作为稳定GPX4的关键上游调节因子,主要通过去除GPX4上K48连接的泛素化来防止其蛋白酶体介导的降解。

图3 USP8去除GPX4上K48残基的K48链泛素化(Ref. Fig4/S4)
结论:本研究在小鼠肠上皮细胞特异性敲除Usp8后,观察到结肠结构改变,小鼠寿命缩短,伴随着肠上皮细胞死亡增加和脂质过氧化迹象增多。功能上,Usp8杂合缺失小鼠可抑制结直肠肿LIU发生和发展,体外抑制USP8可以增加肿LIU细胞对铁死亡的敏感性。机制上,USP8与GPX4发生相互作用,并去除GPX4的多聚泛素化修饰,从而抑制蛋白酶体介导的GPX4蛋白降解。此外,USP8抑制剂联合铁死亡诱导剂能够改善PD-1/PD-L1阻断抗体的治L 效果。这项研究揭示了USP8通过调节GPX4的稳态从而在体内调控铁死亡的机制,为AI症治L 提供了新的治L 靶点。
联系人:
联系手机:
联系电话:
经营模式:
所在地区:
主营项目:
